
A seven-year-old girl in China had never heard her mother's voice. Born with a genetic mutation that silenced her inner ear before she ever left the womb, she lived in a world without birdsong, without laughter, without the particular timbre of someone calling her name. Four months after a single injection into her cochlea, she was holding conversations. Not lip-reading them. Hearing them.
She is one of ten patients in a clinical trial that just delivered something the field of hearing research has chased for decades: a gene therapy that restores hearing in people born profoundly deaf, with one shot and no serious side effects. The results, published in Nature Medicine on April 3, mark a turning point not just for audiology, but for the broader promise of genetic medicine to fix problems that begin before birth.
The Gene That Breaks Silence
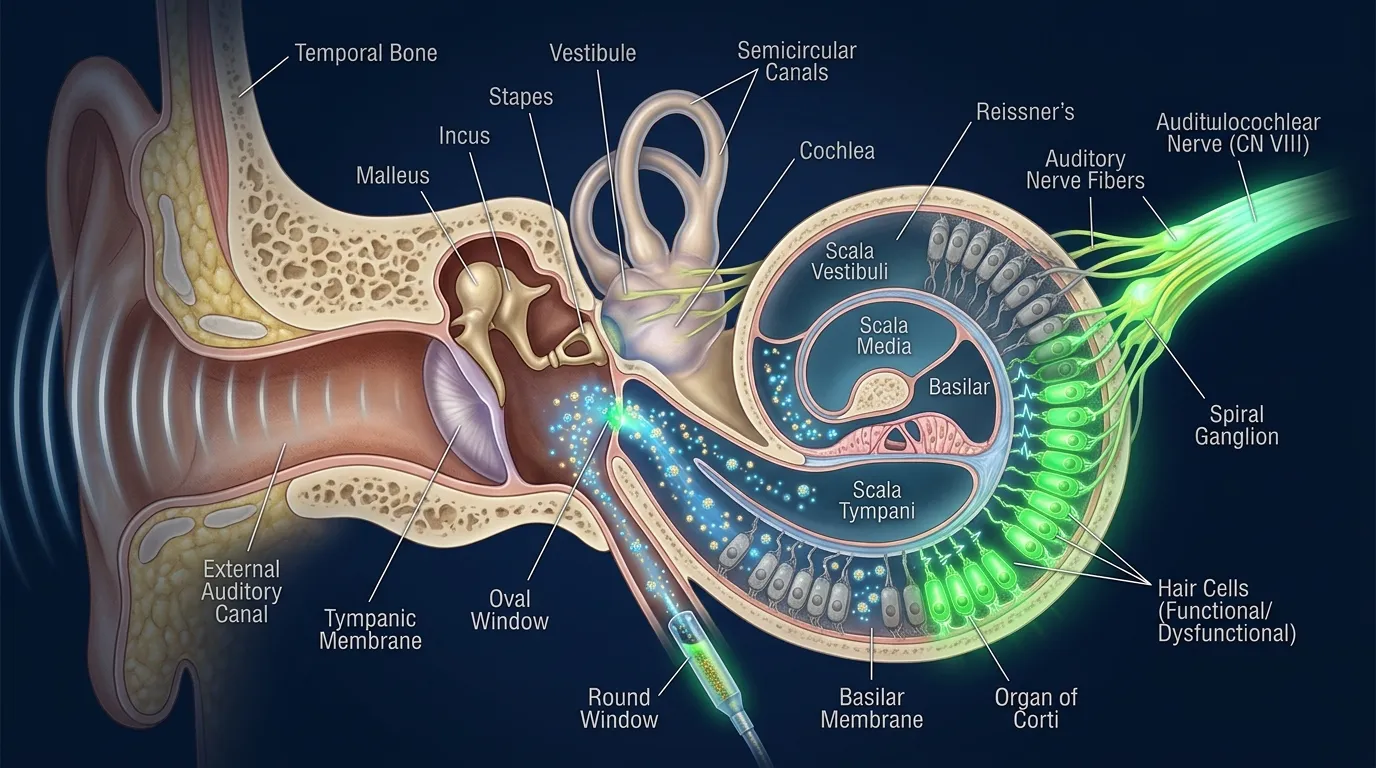
To understand why this therapy works, you need to understand what breaks in the first place. The patients in this trial all carry mutations in a gene called OTOF, which encodes a protein named otoferlin. In a normally functioning ear, sound waves vibrate tiny hair cells inside the cochlea, the snail-shaped structure of the inner ear. Those vibrations need to be converted into electrical signals that travel along the auditory nerve to the brain. Otoferlin is the molecular bridge that makes that conversion happen. It sits at the synapse between hair cells and nerve fibers, packaging neurotransmitters into vesicles and triggering their release every time a hair cell vibrates.
When OTOF is mutated, the hair cells themselves are often perfectly intact. They vibrate in response to sound just as they should. But without functional otoferlin, the signal dies at the synapse. The ear works mechanically, like a telephone with a severed wire. Sound gets in, but the message never reaches the brain. This particular form of deafness, classified as autosomal recessive deafness 9, accounts for roughly 2-8% of congenital hearing loss cases worldwide.
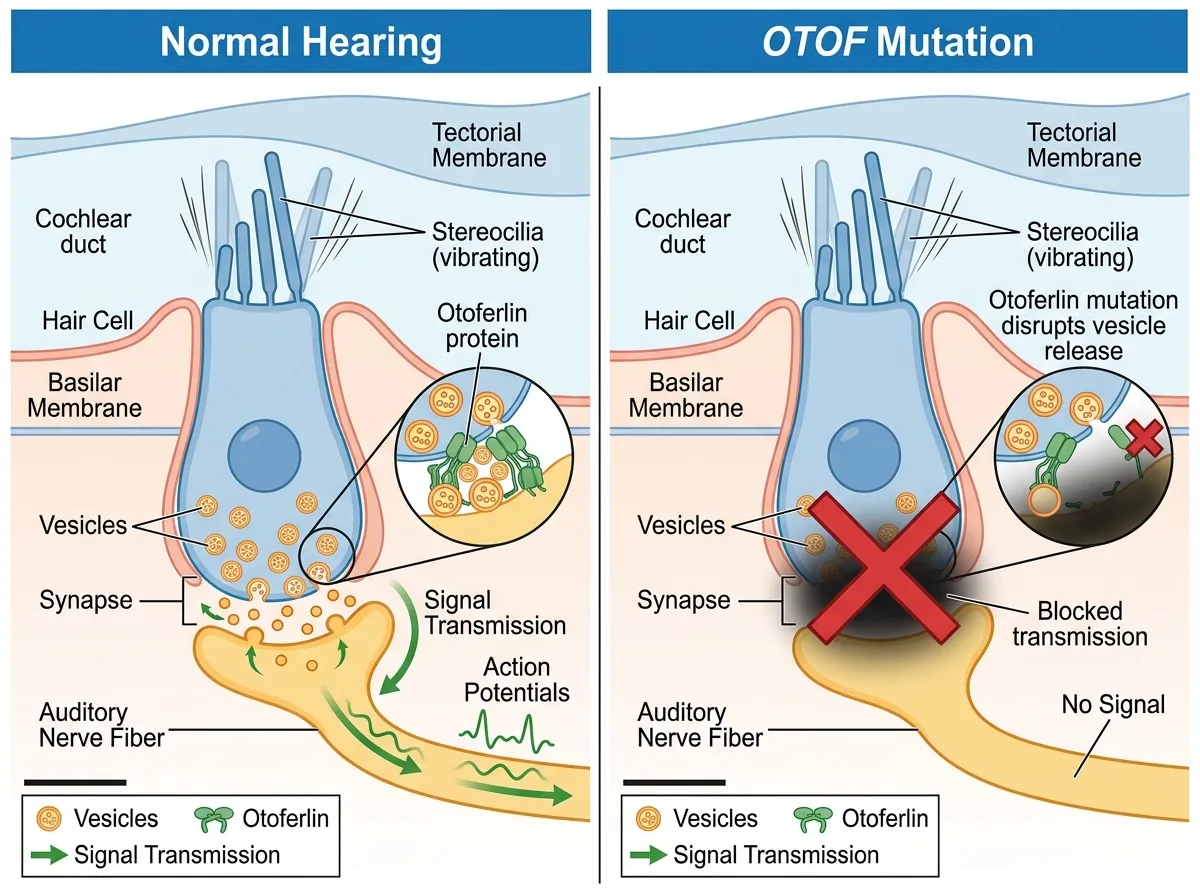
The fact that the structural machinery of the ear remains intact is precisely what makes OTOF deafness a compelling target for gene therapy. You don't need to rebuild the ear. You don't need to regenerate destroyed cells. You just need to deliver the missing protein's blueprint to cells that are already doing most of their job correctly.
One Shot Through the Round Window
The research team, led by Maoli Duan, a consultant and docent at the Department of Clinical Science at Karolinska Institutet in Sweden, conducted the trial across five hospitals in China. The therapy was developed by Otovia Therapeutics Inc. and uses a synthetic adeno-associated virus, or AAV, as a delivery vehicle. The specific capsid, a protein shell that determines which cells the virus can enter, was Anc80L65, a reconstructed ancestral variant designed to efficiently target inner ear cells.
The treatment itself is deceptively simple in concept. A surgeon injects the AAV carrying a functional copy of the OTOF gene through the round window membrane, a thin tissue at the base of the cochlea that provides a natural access point to the inner ear's fluid-filled chambers. The virus particles drift through the cochlear fluid, enter the hair cells, and deliver the genetic instructions for building otoferlin. The cells begin producing the protein on their own.
Ten patients received the injection, ranging in age from 1.5 to 23.9 years. All had confirmed OTOF mutations. All were profoundly deaf before treatment. And all of them, every single one, showed measurable hearing improvement.
What the Numbers Show
The headline result is striking: average pure-tone hearing thresholds improved from 106 decibels (profoundly deaf, unable to hear a jackhammer at close range) to 52 decibels (moderate hearing loss, able to hear normal conversation). Click auditory brainstem responses, which measure how the brain processes sound, improved from 101 to 48 decibels. These are not marginal gains. A shift of 54 decibels moves a person from a world of silence into a world where speech, music, and environmental sounds become accessible.
Most patients showed improvement within a single month. The therapy was well tolerated across the 6-to-12-month follow-up period, with 162 recorded adverse events, all classified as grade I or II (mild to moderate). The most common was a temporary decrease in neutrophil percentage, a type of white blood cell. No serious adverse reactions were observed.
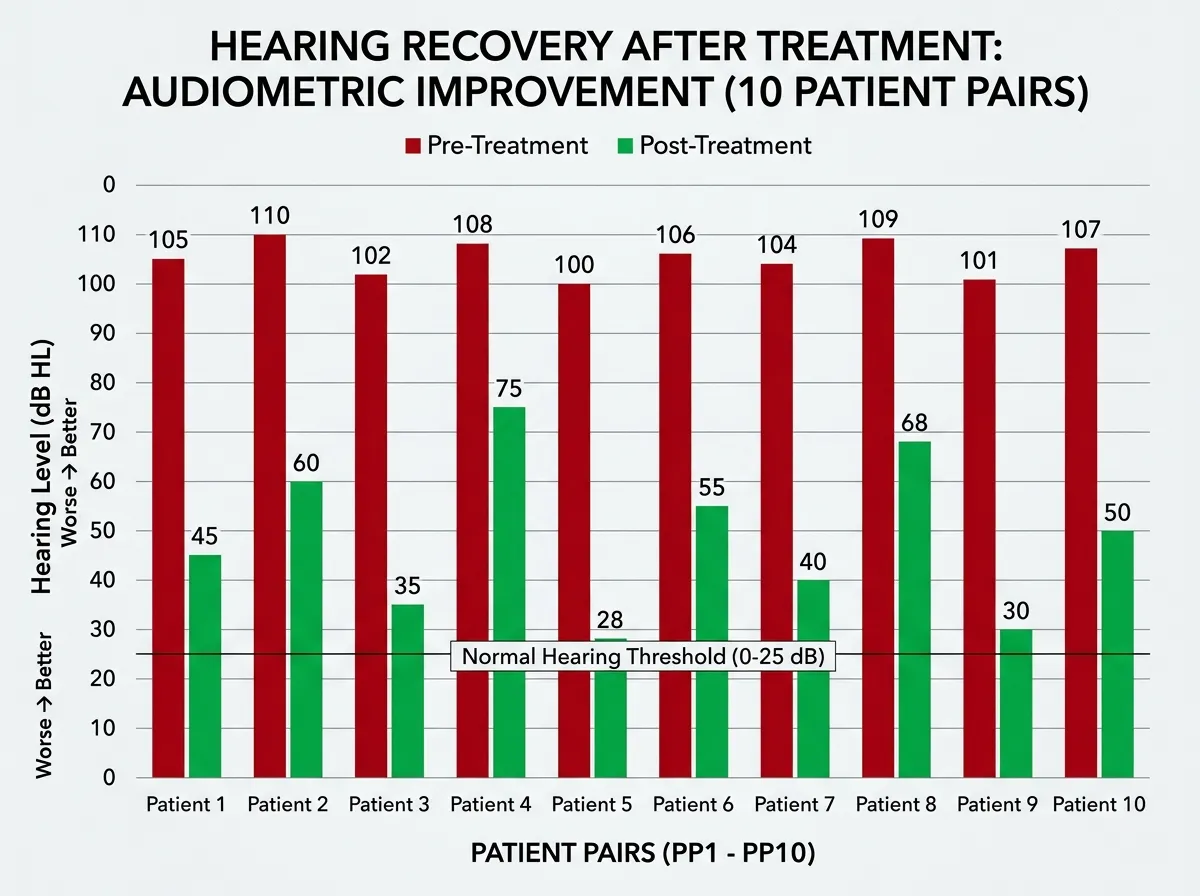
The age effect deserves particular attention. Children between five and eight years old showed the most dramatic responses, consistent with the neuroscience of auditory development. The brain's auditory cortex retains significant plasticity during childhood, meaning it can still learn to process sound signals even after years of silence. The seven-year-old patient who achieved near-normal hearing represents the upper end of what's possible when the therapy arrives during this critical window. The oldest patient, nearly 24, also improved, but the gains were more modest, suggesting that earlier intervention produces better outcomes.
"This is a huge step forward in the genetic treatment of deafness, one that can be life-changing for children and adults," said Maoli Duan, the study's corresponding author.
The Long Road to a Single Shot
This trial didn't emerge from nowhere. The scientific path to OTOF gene therapy stretches back decades, through a series of advances in both genetics and viral vector engineering that made it possible to deliver a large gene to a tiny, fluid-filled structure deep inside the skull.
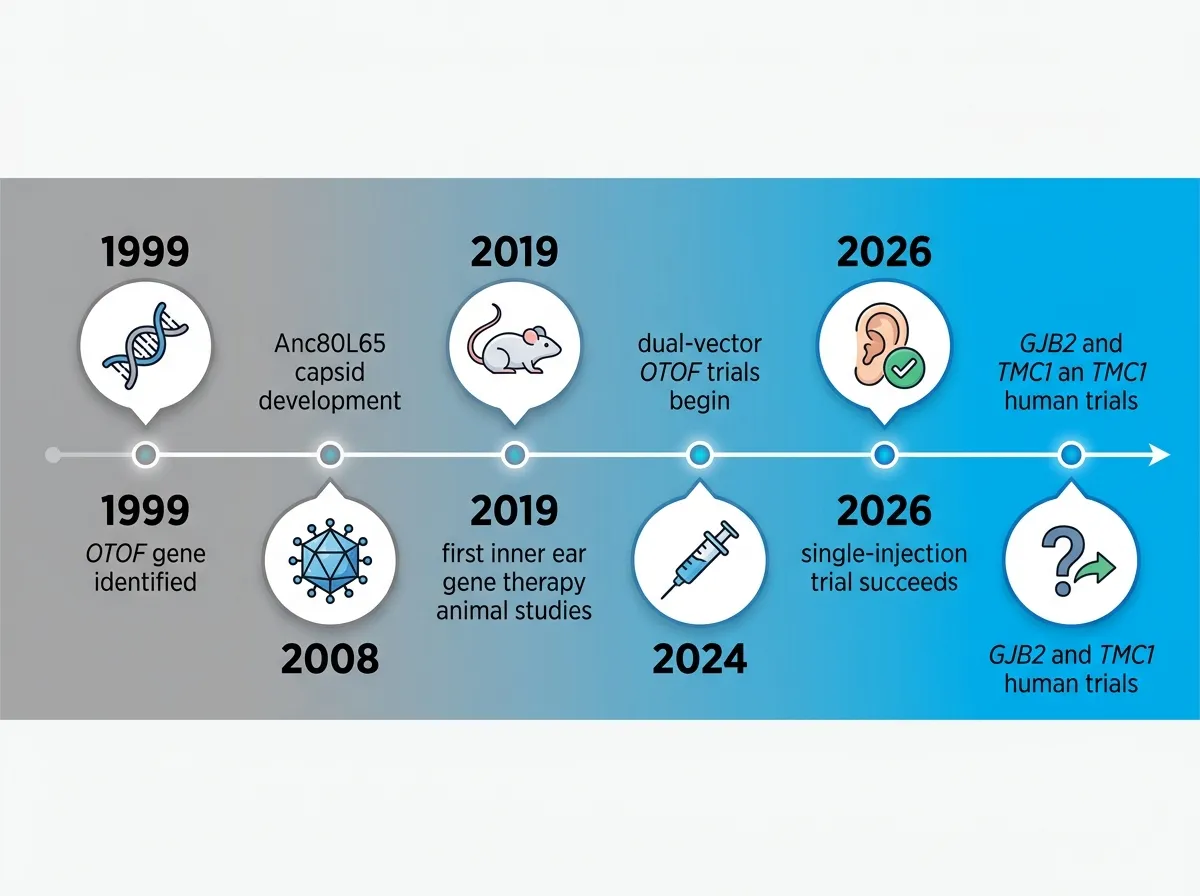
OTOF was identified as a deafness gene in 1999, when researchers linked mutations to a specific form of non-syndromic hearing loss. But knowing which gene was broken and knowing how to fix it were separated by an enormous technical gap. The OTOF gene is unusually large, roughly 6 kilobases of coding sequence, which pushes the carrying capacity of standard AAV vectors. Early gene therapy attempts for other conditions had already established AAV as the safest and most effective delivery platform, but packaging a gene this big required either splitting it across two vectors (a dual-vector approach used in some competing trials) or engineering a capsid efficient enough to deliver a full-length construct.
The Anc80L65 capsid used in this trial represents one solution. Reconstructed computationally from ancestral AAV sequences, it was specifically selected for its ability to transduce cochlear hair cells at high efficiency. Combined with advances in surgical technique for round-window delivery, the platform finally reached the point where a single injection could reliably get enough functional gene copies into enough cells to restore meaningful hearing.
This trajectory mirrors what's happening across regenerative medicine more broadly. CRISPR-based therapies have moved from cutting genes to activating them, and lab-grown organs are being transplanted into children for the first time. The common thread is that decades of foundational research are converging into clinical results almost simultaneously.
Beyond OTOF: The Hearing Genes Waiting in Line
OTOF mutations cause a relatively small fraction of genetic deafness. The larger prizes are GJB2 and TMC1, genes responsible for far more cases of inherited hearing loss worldwide. GJB2 alone accounts for roughly half of all genetic deafness in many populations. Duan's team has already reported promising results in animal studies targeting these genes, and multiple competing groups are racing toward human trials.
The technical challenges differ for each gene. GJB2 encodes connexin 26, a gap junction protein that forms channels between supporting cells in the cochlea. Unlike OTOF deafness, where the hair cells remain intact, GJB2 mutations can cause structural degradation over time, meaning the window for intervention may be narrower. TMC1 presents its own complexities: the protein sits directly in the mechanotransduction channel of hair cells, and restoring it requires precise expression levels to avoid toxicity.
Still, the OTOF trial provides a proof of concept that extends well beyond its specific target. It demonstrates that AAV vectors can safely reach cochlear cells in humans, that gene expression persists for at least a year, and that the brain can learn to process newly restored auditory signals even after years of silence. Those principles apply to any gene therapy targeting the inner ear.
The World Health Organization estimates that 430 million people worldwide have disabling hearing loss, and genetic factors contribute to roughly 50-60% of childhood cases. Cochlear implants, the current standard of care for profound deafness, bypass the damaged biology entirely by electrically stimulating the auditory nerve. They work, but the sound they produce is artificial, limited in frequency range, and requires external hardware. A gene therapy that restores the ear's natural signaling mechanism offers something fundamentally different: biological hearing, produced by the patient's own cells, with no device required.
What Comes Next
The trial was small: ten patients, a single arm, no control group. Larger, randomized studies will need to confirm the results and establish long-term safety beyond the current 12-month follow-up. Questions remain about durability (will the therapy need to be repeated?), optimal timing (how young is too young, and how old is too old?), and whether bilateral treatment, injecting both ears, produces compounding benefits.
But the direction is unmistakable. For the first time, a single injection has taken people from profound deafness to conversational hearing. The seven-year-old girl who can now hear her mother calling her name isn't a statistical anomaly or a best-case-scenario outlier. She's one of ten, and all ten improved. The era of genetic hearing restoration isn't approaching. It's here.
Sources
- Deafness Reversed: One Injection Restores Hearing in Just Weeks - ScienceDaily
- Gene Therapy Restored Hearing in Deaf Patients - Karolinska Institutet
- Gene Therapy Restores Hearing in Those with Deafness - Neuroscience News
- Study: OTOF Gene Therapy Restored Hearing in Deaf Patients - The Hearing Review