
For three decades, the dominant strategy for treating Alzheimer's disease has been the same: clear the amyloid plaques from the brain. Drug after drug has tried. Most have failed entirely. The few that succeeded, like lecanemab (approved in 2023), managed to slow cognitive decline modestly, but the disease kept progressing. The plaques kept forming. Patients kept declining.
A team at Heidelberg University may have found part of the reason. In a study published in Molecular Psychiatry, neurobiologist Prof. Dr. Hilmar Bading and his colleagues identified a toxic partnership between two brain proteins that kills nerve cells, damages synapses, destroys mitochondria, and, in a vicious feedback loop, accelerates the very amyloid buildup that other drugs are trying to remove. They call it the "death complex." And in mice engineered to develop Alzheimer's, they broke it apart.
The Jekyll-and-Hyde Receptor
To understand what Bading's team found, you need to know something strange about one of the brain's most important signaling molecules: the NMDA receptor.
NMDA receptors are proteins that sit on the surface of nerve cells and respond to glutamate, the brain's primary excitatory neurotransmitter. When you form a memory, learn a new skill, or process sensory information, NMDA receptors are doing much of the work. They are essential for cognition. Without them, the brain cannot function.
But NMDA receptors don't all behave the same way. Their behavior depends entirely on where they sit on the neuron. Receptors located at synapses, the junctions where nerve cells communicate with each other, promote cell survival and support learning and memory. These are the good actors. Receptors located outside synapses, in what neuroscientists call "extrasynaptic" positions, can do the opposite. Under certain conditions, they trigger pathways that damage and eventually kill the very neurons they sit on.
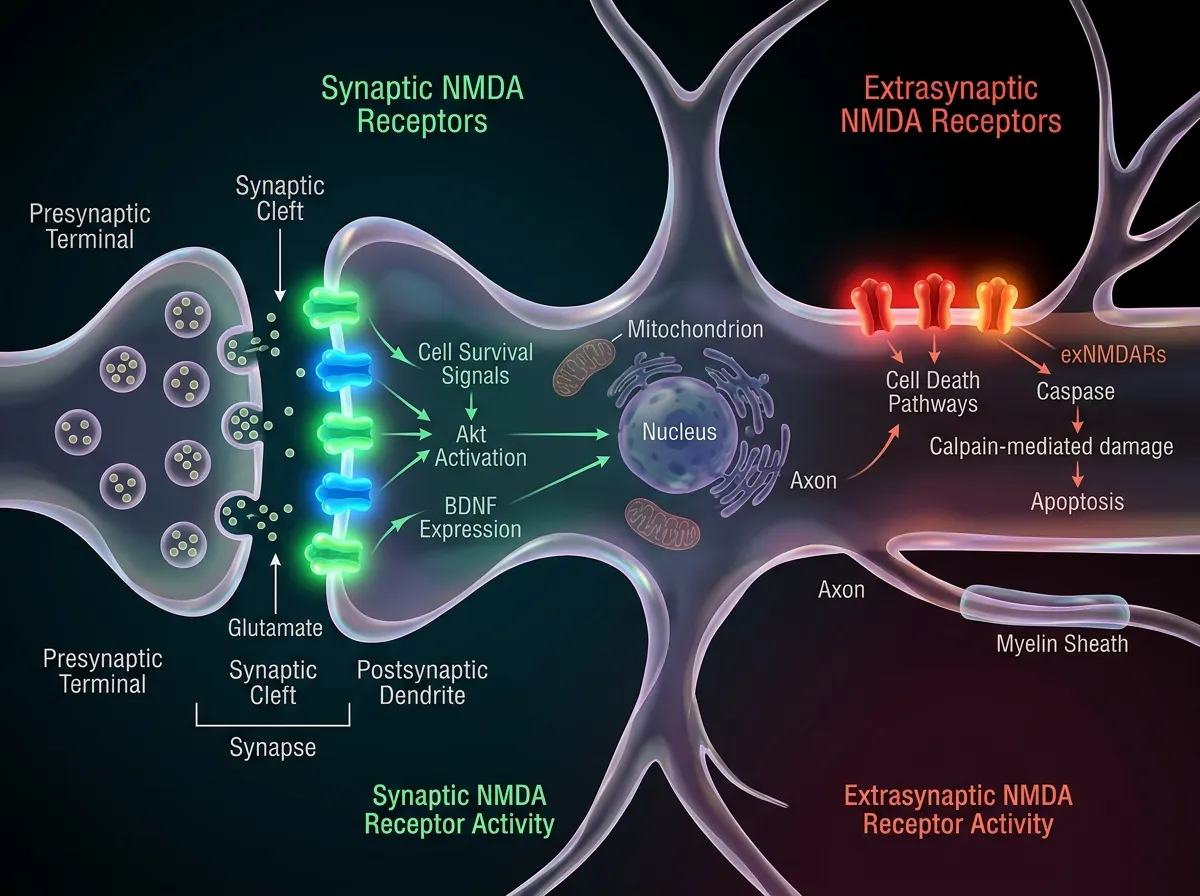
This dual nature has been known since Bading's own earlier work in the 2000s. Synaptic NMDA receptors protect brain cells. Extrasynaptic ones can destroy them. The question that lingered for years was straightforward but unanswered: what determines when extrasynaptic NMDA receptors switch from harmless bystanders to active killers?
When TRPM4 Enters the Picture
The answer, Bading's team discovered, is another protein: an ion channel called TRPM4.
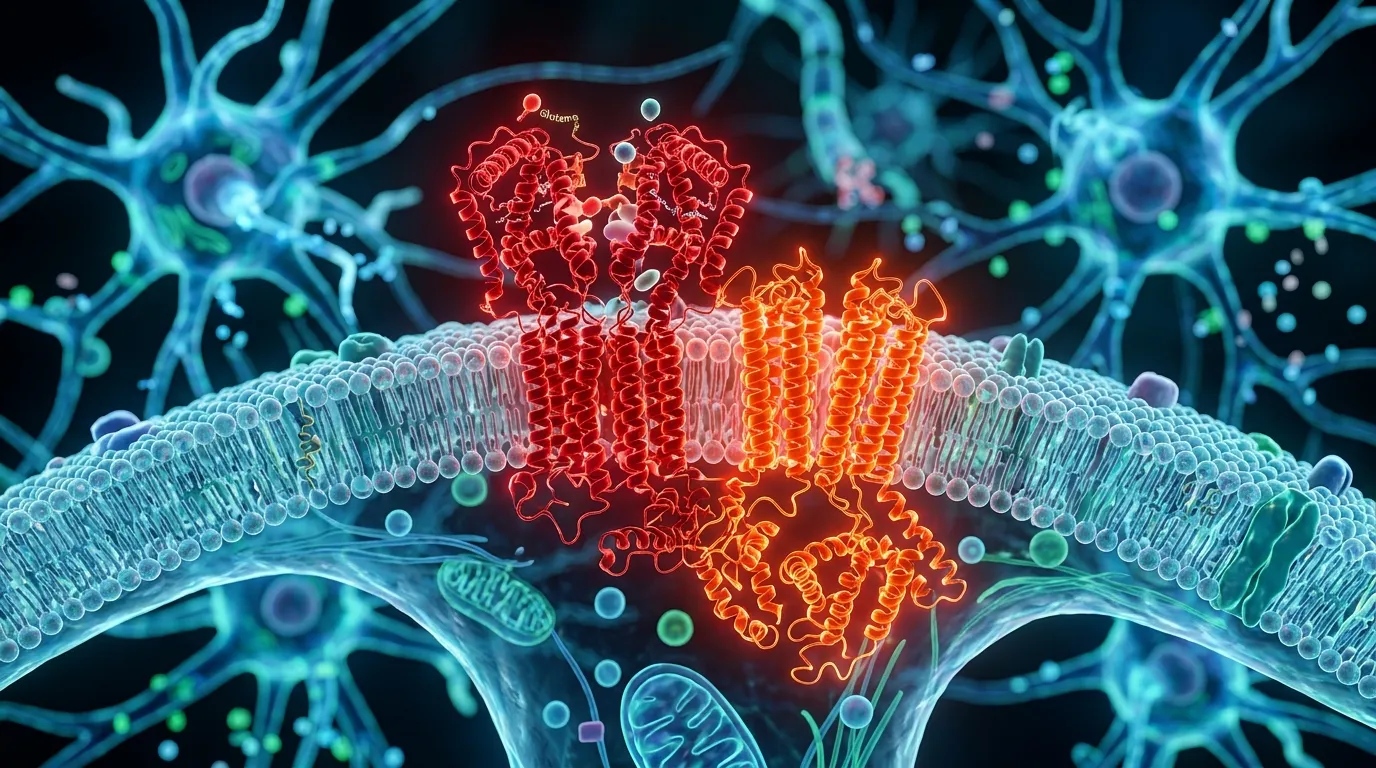
TRPM4 normally has nothing to do with neurodegeneration. It's an ion channel, a protein that controls the flow of charged particles across cell membranes, and it appears in many tissues throughout the body. But when TRPM4 physically binds to an extrasynaptic NMDA receptor, something changes. The receptor's behavior transforms. It begins allowing toxic levels of calcium to flood into the neuron, triggering a cascade of damage: mitochondria swell and break apart, synapses deteriorate, and the cell begins a slow march toward death.
Bading's team found that this NMDAR/TRPM4 complex, the "death complex," exists at significantly elevated levels in the brains of mice engineered with the 5xFAD model of Alzheimer's disease, one of the most widely used laboratory models for the condition. Healthy mouse brains had the complex too, but at much lower levels. In the Alzheimer's mice, something was causing TRPM4 to bind extrasynaptic NMDA receptors at an accelerated rate, creating more death complexes and killing more neurons.
The interaction creates a feedback loop that makes the disease self-reinforcing. As neurons die and synapses deteriorate, the brain's ability to clear amyloid worsens, which in turn promotes more inflammation, more TRPM4 binding, and more cell death. "Instead of targeting the formation or removal of amyloid from the brain, we are blocking a downstream cellular mechanism," Bading explained in a university press release. The complex, he argues, is not just a consequence of Alzheimer's. It is an engine driving the disease forward.
Breaking the Complex Apart
Bading's team didn't just identify the death complex. They had a tool to dismantle it.
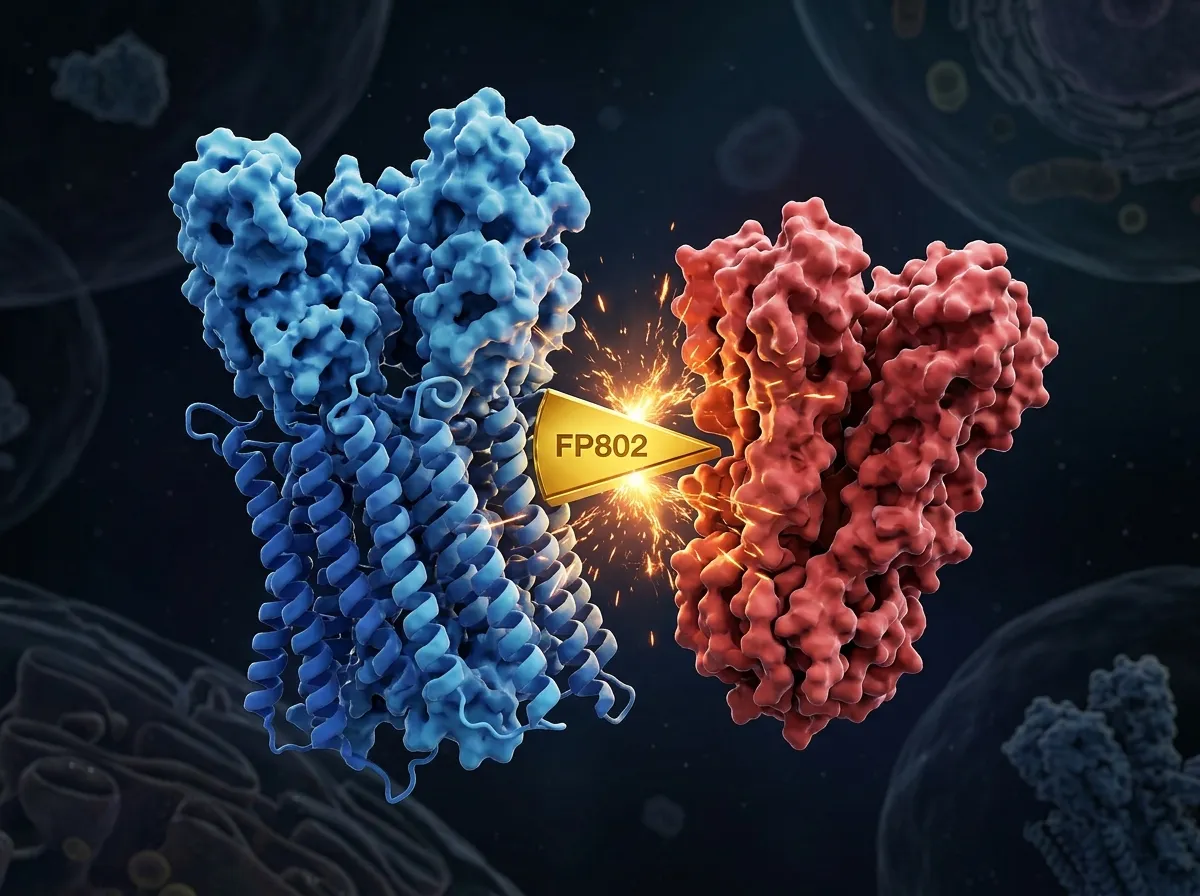
The compound is called FP802, a "TwinF Interface Inhibitor" that Bading's lab developed in earlier research. FP802 works by binding to a specific contact surface, the "TwinF" interface, where TRPM4 physically attaches to NMDA receptors. By occupying that surface, FP802 prevents the two proteins from connecting. Without the connection, the death complex cannot form.
What makes FP802 particularly interesting is its precision. It doesn't block NMDA receptors entirely, which would be catastrophic for cognition. It doesn't interfere with synaptic NMDA receptors at all. It specifically prevents the toxic partnership between TRPM4 and extrasynaptic NMDA receptors, leaving the brain's normal signaling architecture intact.
Dr. Jing Yan, a member of the research team now working at FundaMental Pharma, led much of the experimental work demonstrating FP802's selectivity. The compound can be administered orally, a practical advantage over many experimental neurological drugs that require injection or intravenous delivery.
What the Mice Tell Us
The researchers treated 5xFAD Alzheimer's mice with FP802 and tracked the results across multiple measures of disease progression.
The findings were consistent across every metric they examined. Disease progression slowed markedly. The mice retained their learning and memory abilities at levels far closer to healthy animals than untreated Alzheimer's mice. Synapse loss, one of the earliest and most damaging features of Alzheimer's, was prevented or minimized. Mitochondrial damage, which starves neurons of the energy they need to survive, was likewise limited.
Most strikingly, beta-amyloid deposits, the plaques that have been the focus of Alzheimer's research for decades, were significantly reduced. This was not because FP802 directly targets amyloid. It doesn't. The reduction appears to be an indirect effect of breaking the feedback loop: healthier neurons are better at clearing amyloid, so stopping the death complex slows plaque formation from the cellular level up.
This is a meaningful distinction from drugs that approach brain disease from entirely different angles. Antibody therapies like lecanemab and donanemab use engineered proteins to strip amyloid from the brain directly. They work, to a degree, but the plaques keep reforming because the underlying cellular machinery producing them remains damaged. FP802 addresses that machinery.
Rethinking Three Decades of Amyloid-First Thinking
The "amyloid hypothesis" has dominated Alzheimer's research funding, clinical trials, and drug development since the early 1990s. The idea is straightforward: amyloid plaques accumulate in the brain, they damage neurons, and therefore removing them should stop or slow the disease. Billions of dollars have been invested in this premise.
The results have been humbling. Of the roughly 200 drugs tested in Alzheimer's clinical trials between 2000 and 2022, more than 99% failed. The few that showed efficacy, including lecanemab (marketed as Leqembi) and donanemab (marketed as Kisunla), achieved modest results: slowing cognitive decline by roughly 25-35% over 18 months, while carrying risks of brain swelling and microbleeds. These drugs cost between $26,000 and $32,000 per year. The clinical benefit, while real, has divided the medical community on whether individual patients can notice the difference.
Bading's work suggests a reason for this frustration. If the death complex is an independent driver of neurodegeneration, then clearing amyloid addresses a symptom without fixing the engine. The feedback loop means new plaques will form as long as the NMDAR/TRPM4 complex is still killing neurons and degrading the brain's housekeeping capacity. It also explains why even successful amyloid-clearing drugs produce only modest cognitive benefits: the neurons are still dying through the death complex pathway, just slightly more slowly because the amyloid burden is lighter.
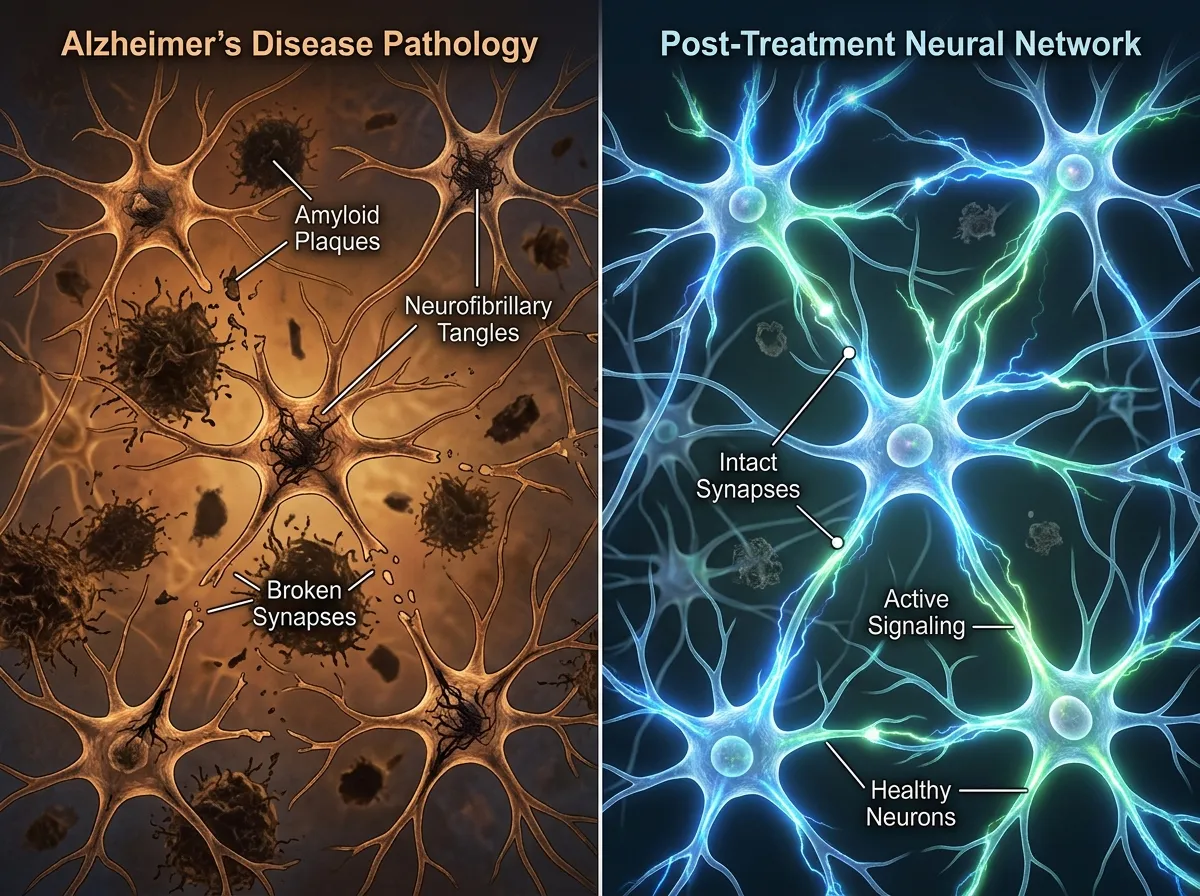
This doesn't mean the amyloid hypothesis is wrong. Amyloid clearly plays a role in the disease. But Bading's research adds a layer that the field has been missing: the cellular mechanism by which neurons actually die may be partially independent of plaque burden, and targeting that mechanism directly could be more effective than targeting plaques alone. A combination approach, pairing amyloid clearance with death complex disruption, could prove more powerful than either strategy in isolation.
The parallel to cancer treatment is instructive. Oncology spent decades focused on killing tumor cells directly through chemotherapy and radiation. Then researchers discovered that the tumor microenvironment, including the surrounding tissue, blood supply, and immune response, mattered just as much as the tumor itself. Modern cancer treatment now combines direct tumor targeting with microenvironment modification. Alzheimer's may eventually require a similar strategic shift, and the death complex could be the target that makes combination therapy viable.
The discovery also resonates with a broader pattern in medical research: diseases once thought to have a single cause turn out to involve unexpected molecular partnerships. The interaction between two otherwise normal proteins, not a mutation, not a toxin, not an infection, is what creates the pathology. This conceptual shift, from targeting individual molecules to targeting specific molecular interactions, could reshape how researchers approach neurodegeneration as a whole.
Where This Leads
Bading is candid about the timeline. "Comprehensive pharmacological development, toxicological experiments, and clinical studies" stand between FP802 and a patient's medicine cabinet, he told Heidelberg University's newsroom. Clinical application remains years away at minimum.
But there are reasons for measured optimism that extend beyond a single compound. The death complex mechanism also appears in animal models of ALS (amyotrophic lateral sclerosis), the motor neuron disease that killed Stephen Hawking and Lou Gehrig. If the NMDAR/TRPM4 interaction is a shared vulnerability across multiple neurodegenerative diseases, a single class of drugs could potentially address conditions that currently have no effective treatment.
FP802's oral bioavailability is another practical advantage. Many experimental neurological drugs fail not because they don't work, but because they can't cross the blood-brain barrier efficiently or require intravenous infusions that limit patient access. An oral compound that reaches the brain effectively would be far easier to deploy at scale, joining a growing list of medical breakthroughs designed for real-world patient delivery.
Roughly 55 million people worldwide live with dementia, a number the World Health Organization projects will reach 139 million by 2050 as populations age. The death complex won't be the whole answer to a disease this sprawling and stubborn. But if Bading is right that the field has been aiming at a downstream symptom while the engine runs unchecked, his team may have identified the target that makes other treatments work better, and the molecular switch that finally lets researchers turn the engine off.
Sources
- Key Mechanism for Alzheimer's Disease Discovered - Heidelberg University
- Scientists discover Alzheimer's hidden "death switch" in the brain - ScienceDaily
- Blocking a Brain "Death Complex" Slows Alzheimer's - Neuroscience News
- The NMDAR/TRPM4 death complex is a major promoter of disease progression in the 5xFAD mouse model of Alzheimer's disease - Molecular Psychiatry